近日,基础医学院李锴、韩晓团队在Cell Reports在线发表了题为 Ets1-dependent Mek-Erk signaling drives adipose tissue macrophage anti-inflammatory polarization to ameliorates insulin resistance的研究论文。该研究发现,转录因子Ets1可作为Mek-Erk信号通路中一个关键的效应器,特异性响应IL-4信号,驱动巨噬细胞向抗炎M2表型极化,从而缓解脂肪组织炎症和胰岛素抵抗。
肥胖已成为全球性的公共卫生挑战。脂肪组织中的慢性低度炎症是导致胰岛素抵抗和2型糖尿病的关键驱动因素。脂肪组织巨噬细胞(ATMs)是脂肪组织中数量最多的免疫细胞,在肥胖状态下,其表型从抗炎的M2型向促炎的M1型极化,加剧代谢紊乱。因此,调控ATMs的M2/M1平衡被认为是改善肥胖相关胰岛素抵抗的潜在策略。
巨噬细胞的M2/M1极化受到多种信号通路的精密调控,其中丝裂原活化蛋白激酶(MAPK)家族的Mek-Erk信号通路在巨噬细胞极化中发挥着复杂而矛盾的作用:一方面,M2极化需要Mek-Erk活性;另一方面,促炎刺激(如LPS)同样激活Erk。临床研究也发现,使用MEK/ERK抑制剂(如trametinib、cobimetinib)在不同研究中表现出截然相反的免疫调节效应。这种功能分歧的根源在于Erk下游效应器的多样性,但其具体机制尚不明确。
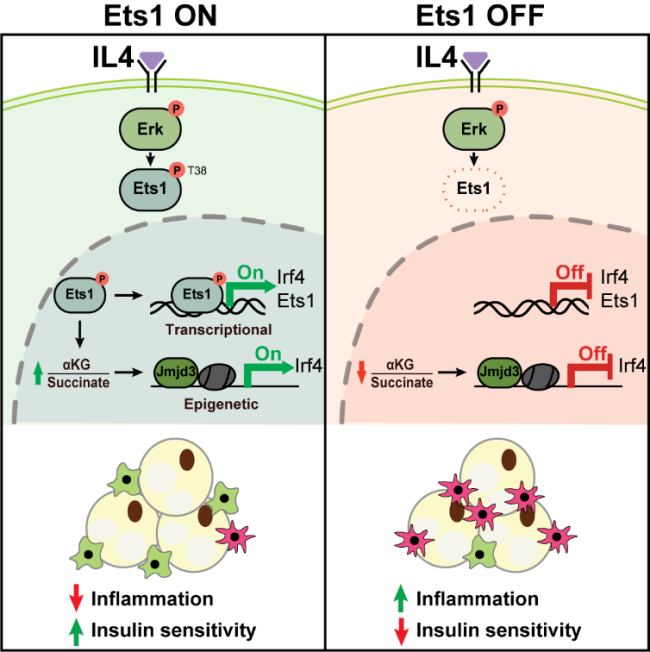
研究团队通过对比IL-4(M2极化诱导因子)与LPS/棕榈酸(M1极化诱导因子)对巨噬细胞的作用后发现:尽管不同刺激均能激活Erk磷酸化,但只有IL-4能够以Mek-Erk信号依赖的方式显著上调Ets1蛋白水平——即Ets1是Mek-Erk通路响应上游信号的差异化调控节点。课题组进一步证实Ets1缺失会加剧高脂饮食诱导的脂肪组织炎症和胰岛素抵抗。本研究发现了Mek-Erk信号通路中一个关键的调控机制:Erk可通过激活特异性下游效应器Ets1,驱动巨噬细胞抗炎极化,从而缓解脂肪组织炎症和胰岛素抵抗。这为理解Erk信号的免疫代谢调控功能提供了新视角,也为肥胖相关代谢性疾病提供了潜在干预靶点。
我校基础医学院李锴、韩晓,第一附属医院刘威及附属泰州人民医院杨淑芳为共同通讯作者,基础医学院硕士生宋吉媛、倪嘉豪及本科生谢坤鑫为该论文的共同第一作者。该研究得到了国家自然科学基金等项目的资助。
原文链接:
https://doi.org/10.1016/j.celrep.2026.112744
(供稿/李锴课题组;审核/王觉进)
